About GCGD
GCGD is an integrated genomics and genetics database for grass carp ( Ctenopharyngodon idellus ), which has been intensively studied due to its important economic value. The annotated genome of a female grass carp, as well as other data from studies on transcriptomes and a genetic linkage map, can be accessed quickly and easily. Some tools have been integrated to facilitate the search and analysis of data, e.g., JBrowse for navigation of genome annotations, BLAST for alignment of similar sequences, EC2KEGG for comparison of metabolic pathways, IDConvert for conversion of IDs among various public databases, and ReadContigs for extraction of sequences from the grass carp genome.
Genetic Map
Genetic Map -- a consensus linkage map constructed with microsatellites and SNPs
General information
A linkage map is an essential framework for mapping traits of interests and is often the first step towards understanding genome evolution. We collected the source for the genetic linkage maps of grass carp in GCGD from Xia et al (2010, BMC Genomics) to help QTL analysis and marker-assisted selection (MAS) breeding programs for economically important traits in grass carp.
Utilization
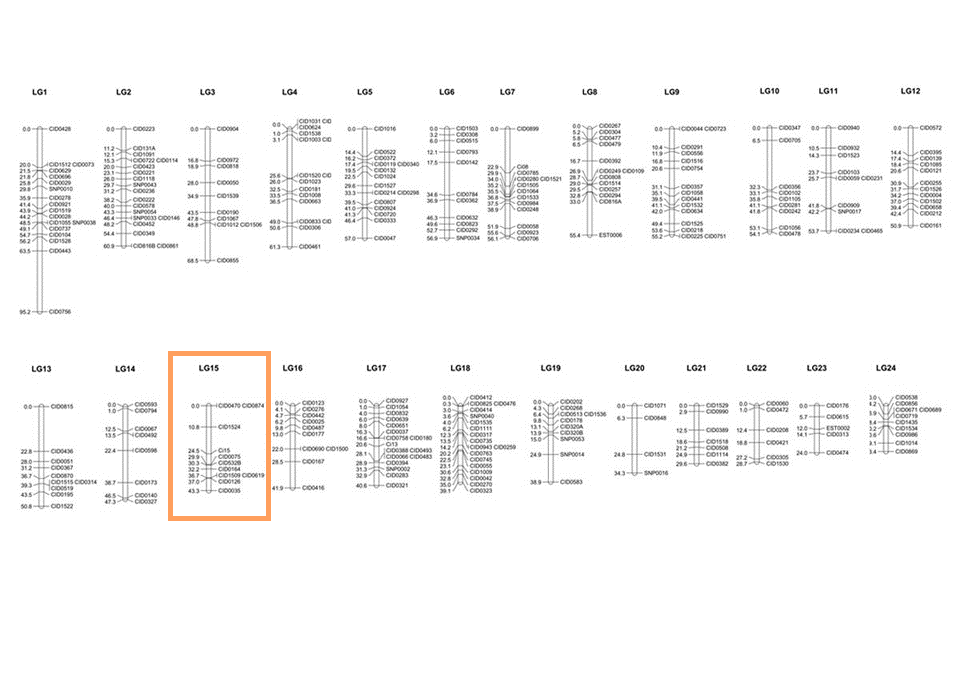
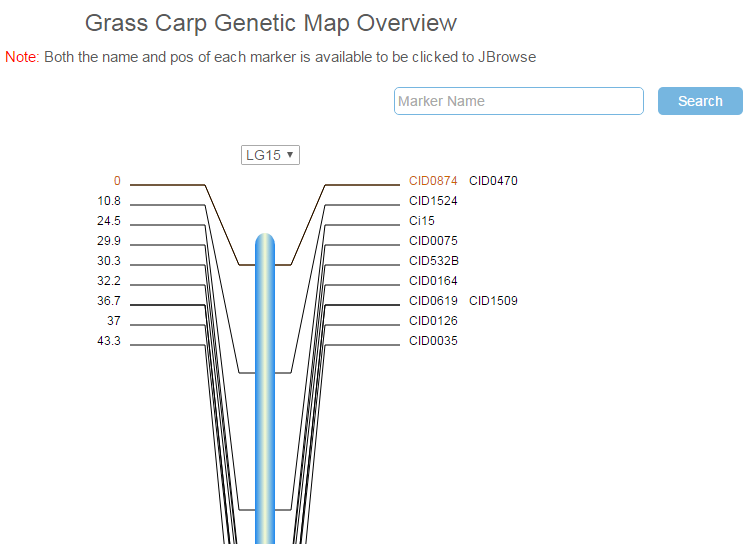
A total of 279 makers (16 SNPs and 263 microsatellite markers) were mapped to 24 linkage groups (LGs) using MapChart, and estimates of map distances between markers were indicated in Kosambi centimorgans. In the page "Genetic Map”, clicking on the chart of any specific linkage group brings a map in SVG (Scalable Vector Graphics) format with marker names / genetic distances on either side as shown below. A drop-down menu is added above the map for switching to other linkage groups. An entry is provided for searching a marker's location and the position of the marker is highlighted with red color. Additionally, the details of a marker can be displayed in JBrowse by clicking on its name or genetic distance.
Note
Some makers are not available to be visualized in JBrowse, because these markers either don't have GenBank number or fail to be aligned to reference genome.
Flowchart
JBrowse
JBrowse -- grass carp genome visualization
General information
JBrowse, a web-based browser developed by Skinner et al. (2009), has been integrated into many genome databases for genome visualization and analysis, such as rosaceae, candidagenome, and pseudomonas. To date, the draft genome, predicted genes, microsatellite markers, and three transcriptome datasets, are available in JBrowse on GCGD. Please refer to the JBrowse page for more information.
Utilization
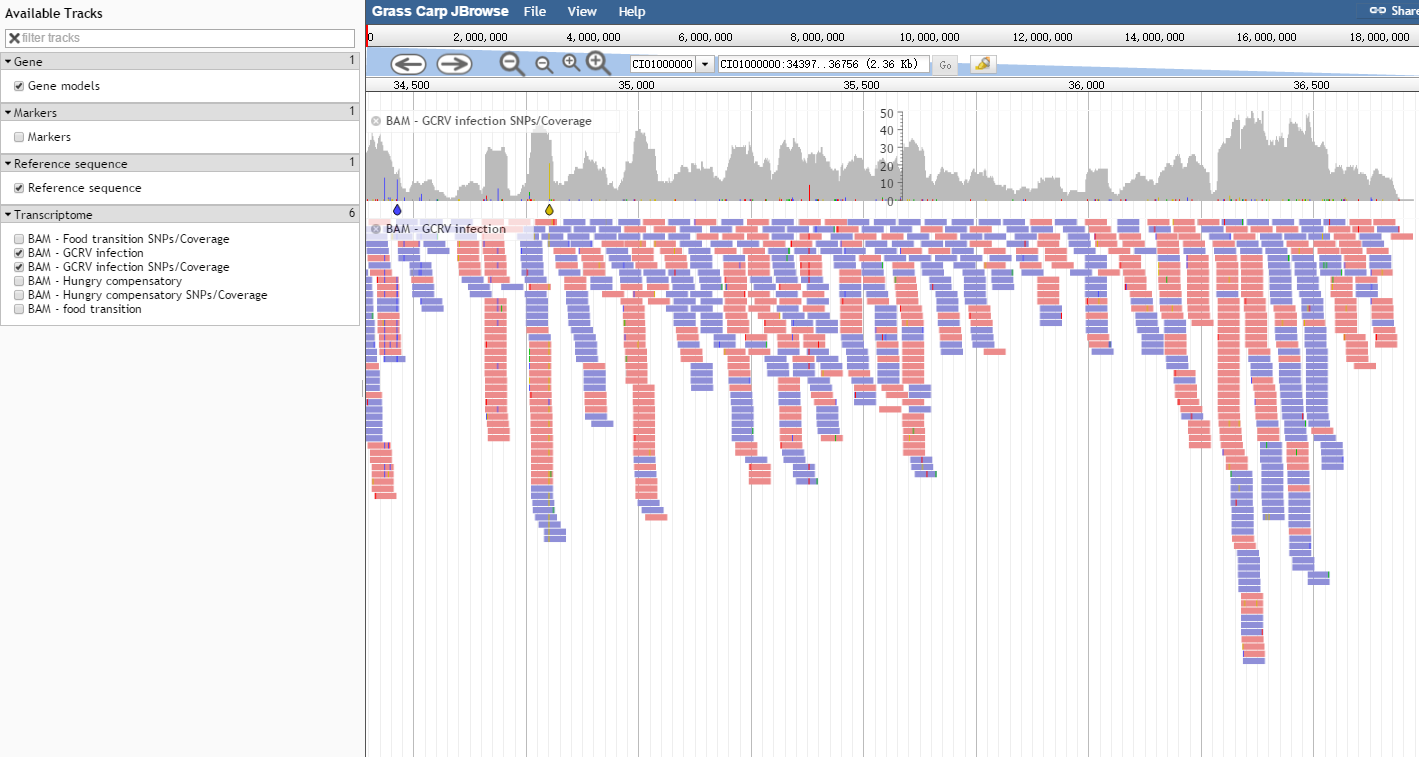
The user interface of JBrowse is shown in Fig. 1. A hierarchical track selector is available in the left-hand panel for choosing features to be displayed. From top to bottom in the main panel, there are menu bar, location bar, navigation bar, and canvas for feature tracks in order. The navigation barprovides buttons for panning/zooming/highlight, a selector for select a reference sequence from the first 40 scaffolds, a text navigation box enabling navigation to features by name, and an overview bar showing the global location of the zoomed-in region.
Fig. 1 The user interface of JBrowse
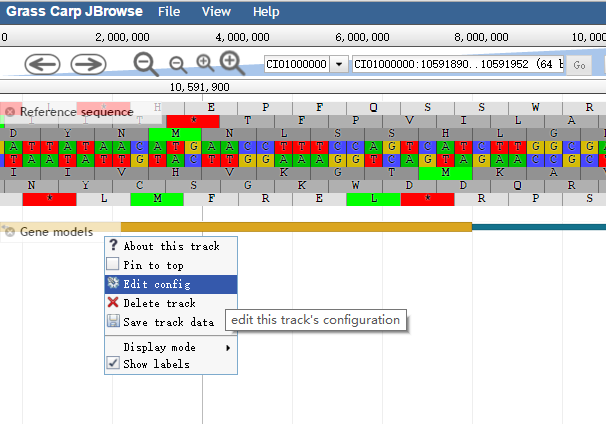
The track "Gene Structure" is displayed in the feature canvas (Fig. 2). Such a track also has its own menu available as a dropdown at the side of the track label. The track menu offers several operations: to display track metadata (the "About" this track option), to pin the track to the top, to edit the track configuration directly, to export the track data in BED or GFF3 format, to delete the track, and so on.
Fig. 2 A "Gene Structure” track
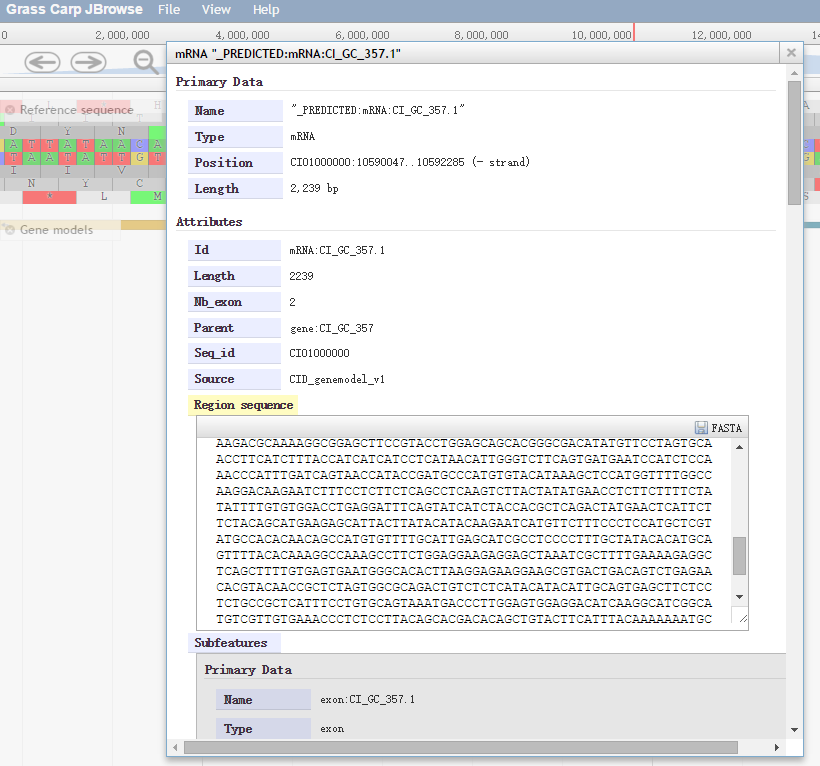
Right-clicking on a feature in a track brings up a context menu, which has two options by default: "View details" and "Highlight feature". Left-clicking on the feature goes directly to the "View details" pop-up window, which lists attributes of the gene. Details of a predicted protein-coding gene include gene ID/name/alias, location, sequence length, ontology terms, and other gene attributes(Fig. 3).
Fig. 3 Gene Structure
Note
JBrowse works well with most browsers (e.g., Chrome, Firefox), and it may not work properly with some versions of Internet Explorer (IE).
SSR
SSR -- Simple Sequence Repeats
General information
SSRs, a small segment of DNA, have usually 1 to 6 bp in length that repeats itself a number of times, and are often referred to as microsatellites. Because of their advantages in the feature of codominance, high polymorphism and high repeatability, SSRs are broadly utilized for species or variety identification, DNA fingerprint profile identification, gene mapping and gene locating, linkage map construction and molecular marker assisted breeding. To facilitate retrieval of SSRs, an in-house python script was used for a genome-wide scanning and more than 6,947,000 SSRs were identified. The overall proportions of different types of SSRs are shown as pie charts in the "Data statistics" page. As for the classification criteria of SSRs, please browse the "Classification criteria" paragraph in the “SSR” page.
Utilization
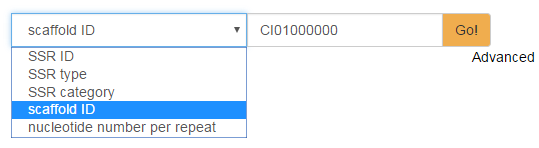
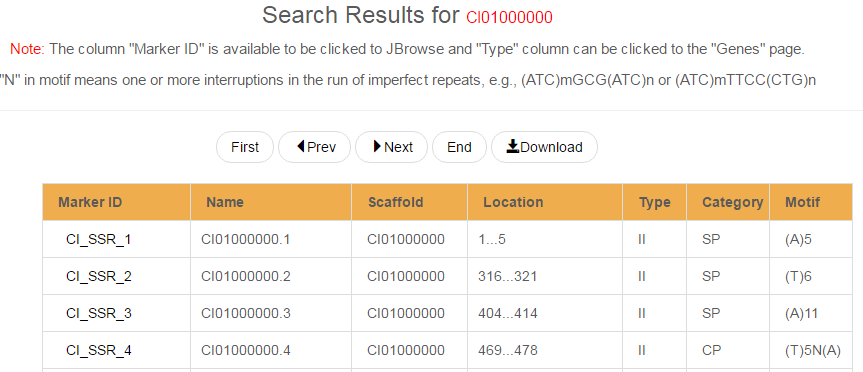
The user can search SSRs quickly in the keyword search entry. As shown in the following figure, the keyword can be a SSR ID, a SSR type, a SSR category, a scaffold ID, or even the nucleotide number per repeat. As for advanced search for SSRs, more keywords can be combined using three logical operators (AND, OR and NOT) to narrow the search results. Searching the keyword “CI01000000” as a "scaffold ID", the results were displayed with detailed information of SSRs, such as category, location, etc.
Flowchart
Gene
General information
So far, 32,811 protein-coding genes have been predicted with function annotations (e.g., GO, KO, EC, KEGG map) and ID mapping to five databases (e.g., NCBI, Ensembl, ZFIN). A summary of the different functional annotations of these genes was shown in the page "Summary of gene annotation" . A total of 1,579 noncoding genes (e.g., snoRNA, snRNA, tRNA, rRNA) have been predicted with locations on the genome. Please visit the page "Download Data" to download the corresponding resources.
Utilization
There are two ways to search for genes in GCGD: (1) Keyword Search. (2) Advanced Search.
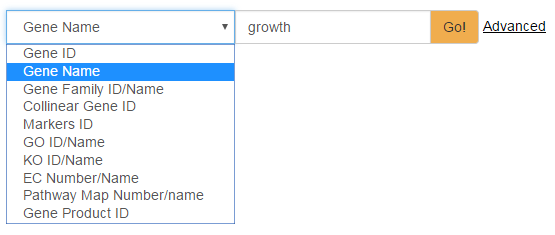
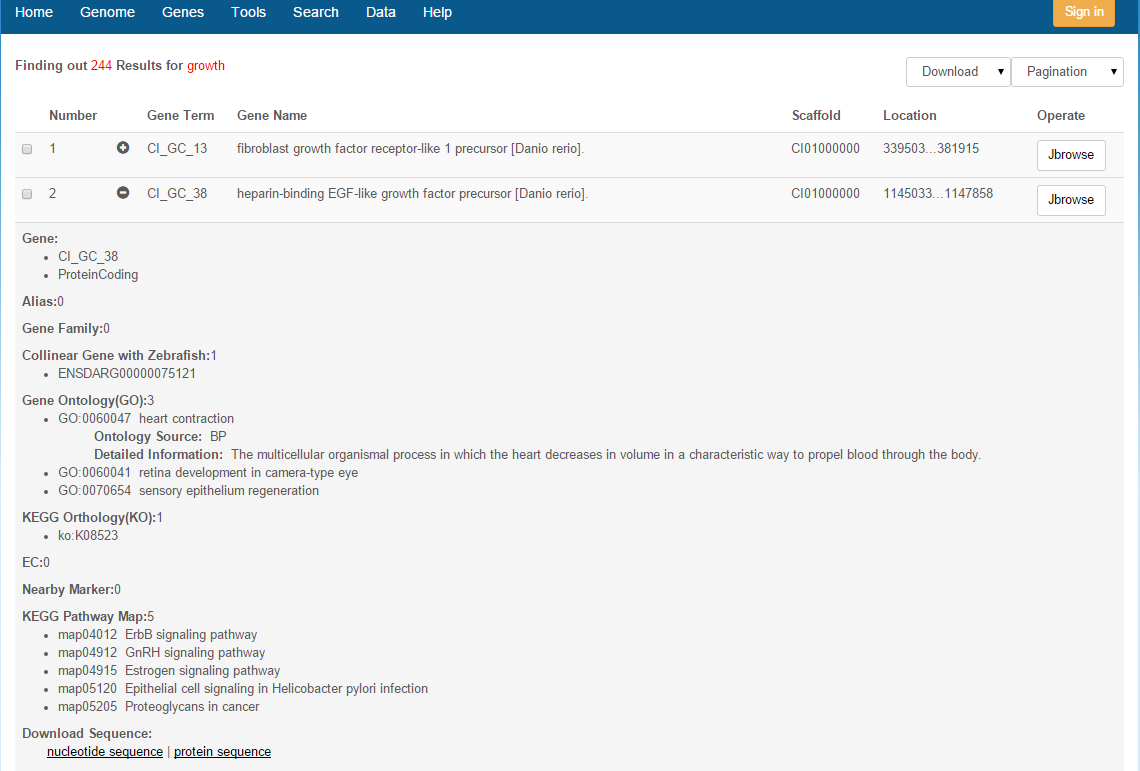
Keyword Search: Valid keywords are Gene ID, Gene name, Gene Family ID/name, Collinear Gene ID, Markers ID, GO ID/Name, KO ID/Name, EC Number/Name, Pathway Map Number/Name, and Gene Product ID (Fig. 1). As an example, taken the Gene ID “CI_GC_1” as the keyword, the results are displayed in a tabular form with fields including gene ID, gene name, scaffold and location. Clicking on the "+" icon in front of a gene ID will bring more details about the gene: ID, name/alias, gene type, protein term, gene family, collinear genes with zebrafish, KO number, GO term, EC number, nearby markers, KEGG pathway map, nucleic acid sequence and amino acid sequence. In addition, Markers nearby gene and gene can be clicked on and be viewed in JBrowse. More detail on genes is given in the "Genes" page.
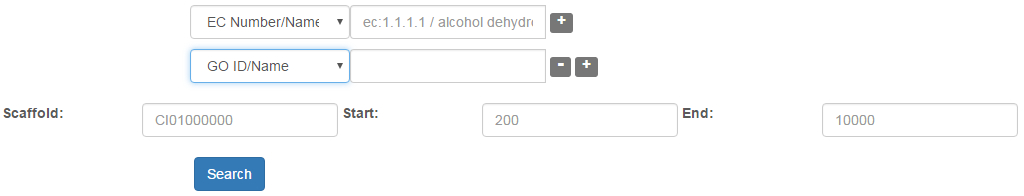
Advanced search: The "Advanced" hyperlink in any page header or in the page "Genes" switches to new searching page a new searching page, in which genes can be screened by more criteria in relation to EC number/name, GO ID/name, KO ID/name, KEGG pathway map number/name, gene family ID/name, collinear gene ID, and species. The criteria for searching could be added or or be removed by clicking on the "+" or "-" buttons in the page (Fig. 2).
Flowchart
Fig. 1 Keyword search
Fig. 2 Advanced search

Gene Family
General information
A gene family is a group of genes that share important characteristics. Classifying individual genes into families helps to describe how genes are related to each other. Annotated genes in GCGD cover 9379 gene families, which can be searched by gene family ID/name from TreeFam database.
Utilization
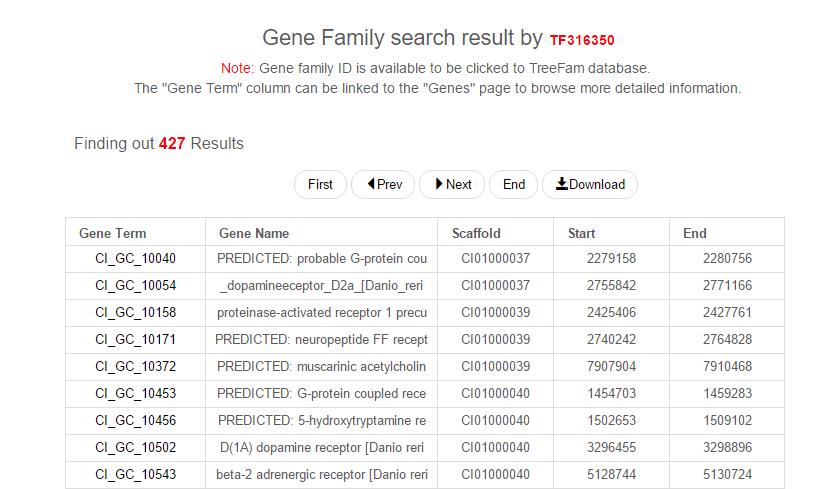
As shown in the Fig. 1, searching for gene family ID "TF316350" resulted in all gene members of this gene family. Each column in the table is Gene ID, Gene Name, Scaffold, Start and End, separately. Additionally, the gene family ID in the title is hyperlinked to the TreeFam database, and every gene ID in the table is linked to a new table as the one returned from the page "Genes". Please visit the page "Gene Family" for more details.
Flowchart
Fig. 1 Gene famaily search

Gene Collinearity
Gene Collinearity -- Gene collinearity between zebrafish and grass carp
General information
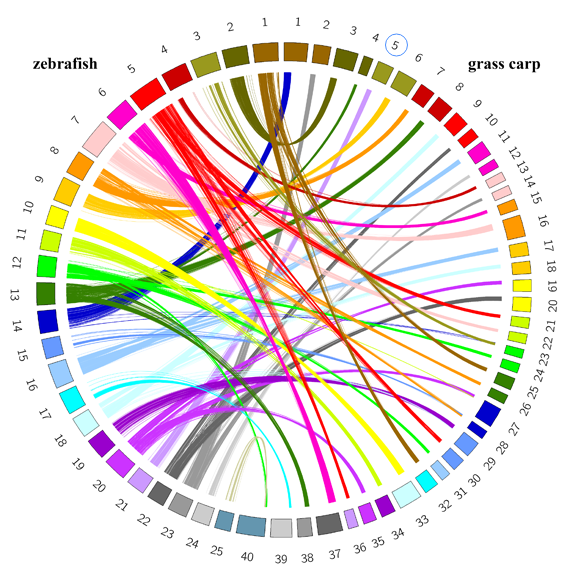
Since both the grass carp and the zebrafish belong to the Cyprinidae family, the relationship of gene collinearity between them helps for chromosome assembly, gene structure analysis and function annotation. To date, 114 scaffolds (573 MB in total) are anchored on 24 linkage groups. Considering that the assembly of the grass carp genome is still at a stage as a "draft" and there are no counterparts for the 25 chromosomes of the zebrafish, we picked up 40 scaffolds with most genes shared with zebrafish to illustrate their gene collinearity. Circos is used for visualization of the relationship of gene collinearity between them.
Utilization
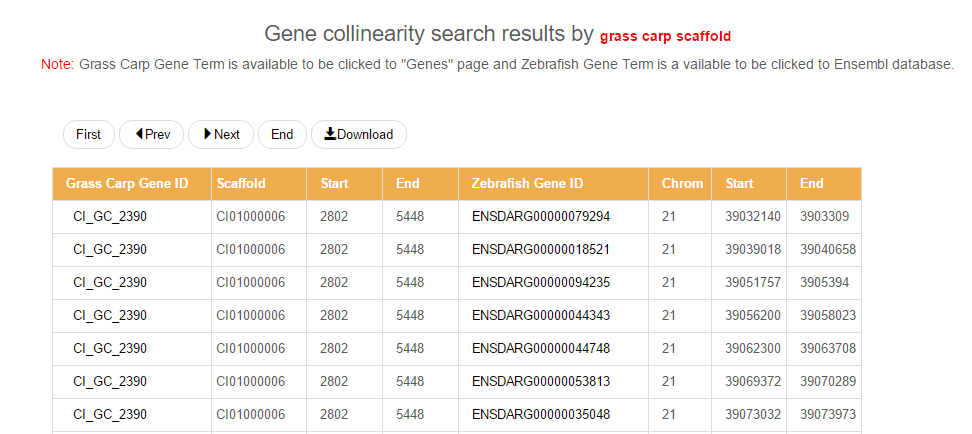
By clicking on any number on the circular graph, all collinear genes can be listed in a table, in which each Gene ID of the grass carp is hyperlinked to a more informative "Genes" page, while every zebrafish Gene ID is linked to the Ensembl database for more details about the gene. See the page "GC-ZF" for details.
Flowchart
BLAST
Blast -- a tool for alignment of similar sequences
General information
BLAST stands for Basic Local Alignment Search Tool and was developed by Altschul et al. (1990) and significantly improved by Altschul et al. (1997) . As one of the most common and useful tools, it has been widely integrated into various databases for searching sequences, such as CarpBase, ZFIN, Ensembl, etc. GCGD also employed BLAST facility for this purpose.
BLAST methods
The NCBI BLAST family of programs includes:
- blastp: Compares an amino acid query sequence against a protein sequence database.
- blastn: Compares a nucleotide query sequence against a nucleotide sequence Database.
- blastx: Compares a nucleotide query sequence translated in all reading frames against a protein sequence database.
- tblastn: Compares a protein query sequence against a nucleotide sequence database dynamically translated in all reading frames.
- tblastx: Compares the six-frame translations of a nucleotide query sequence against the six-frame translations of a nucleotide sequence database.
Databases
- Grass carp genome sequences
- Grass carp protein sequences
- Zebrafish (Danio rerio) genome sequences (genome version Zv9)
- Zebrafish protein sequences (protein version Zv9)
Running BLAST
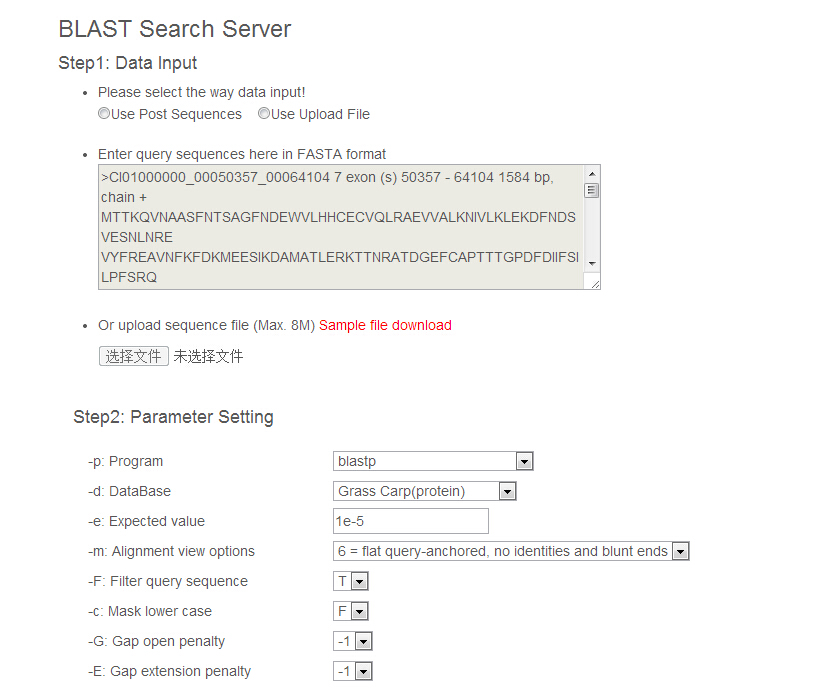
The operations for BLAST mainly comprise of two steps. The first step is to upload query sequences in FASTA format, which can either be pasted in a text box or be uploaded as a local file. The second step is to set a series of parameters, especially, select the appropriate blast program and dataset you want to query.
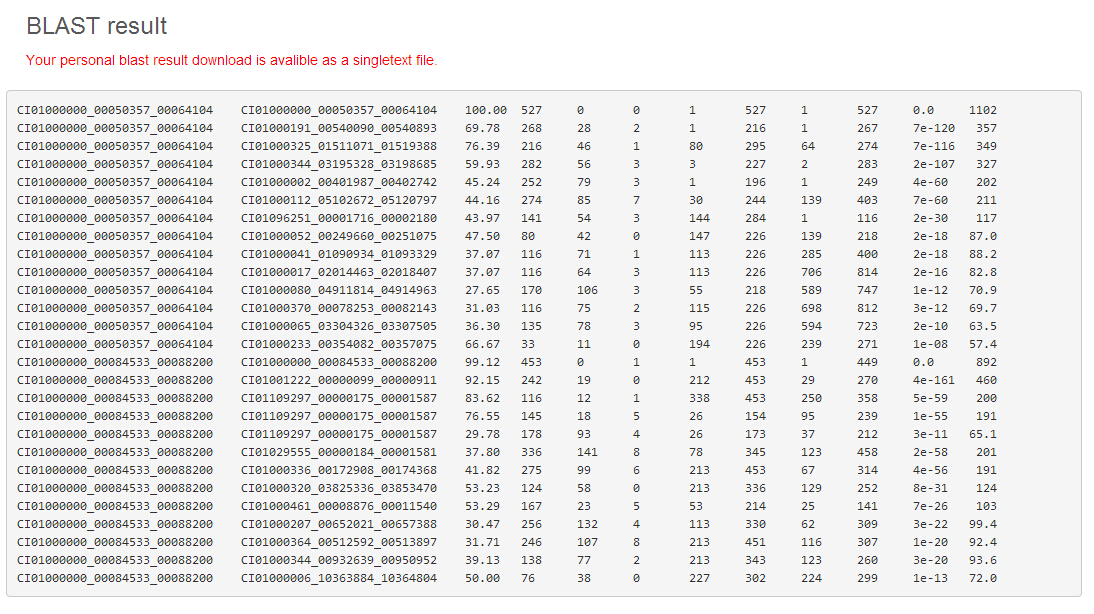
Results Summary
A BLAST request will run immediately and return results directly to the user's web browser. Additionally, a button helps to download the result file. To implement sequence similarity search, please jump to the BLAST page.
Flowchart
ReadContigs
ReadContigs -- an in-house python script for extraction of sequences from grass carp genome
General information
An in-house python script, ReadContigs, is integrated into GCGD to allow the user to obtain grass carp genome sequence of interest.
Utilization
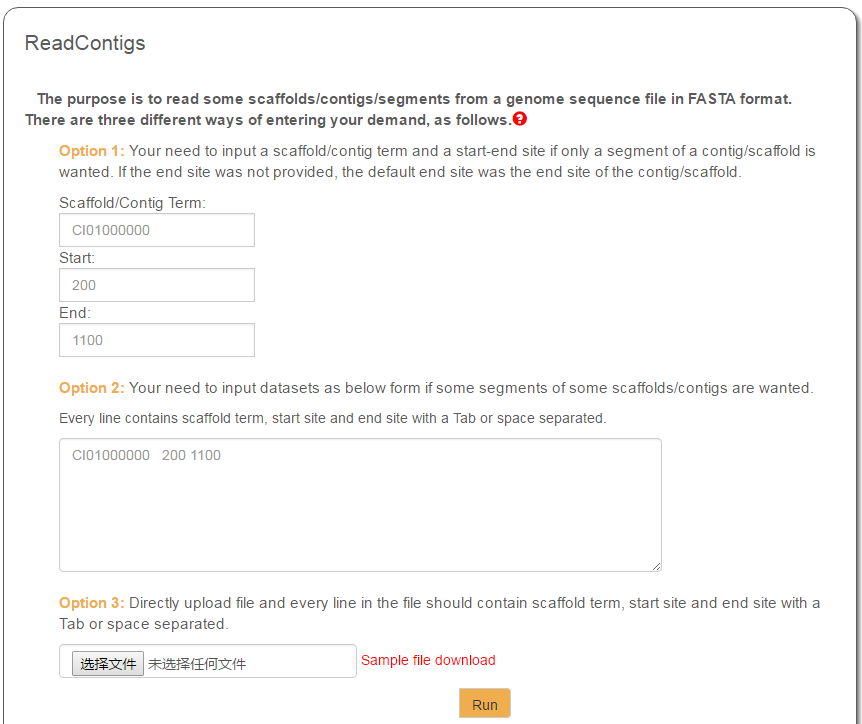
GCGD provides three different options for data input: (1) input a scaffold/contig ID and a start/end site, (2) directly input a query list in the text entry, and (3) upload a query list as a local file. With scaffold/contig IDs and sequence location information, requested sequences can be extracted and returned.
Note
Every query you enter should be composed of scaffold/contig ID and sequence location (start-end site). Take a file as example. Every row in the file contains three columns (scaffold ID, start site and end site) separated by TAB characters or SPACE characters.
Flowchart
IDConvert
IDConvert -- an in-house python script for the conversion of IDs across databases
General information
In the era of big Data, the exchange and connection of information among databases is particularly important for gene function research. An in-house python script named as IDConvert is used for ID mutual conversion between GCGD and four other databases: NCBI, Ensembl, ZFIN and Uniprot/Swiss-Prot.
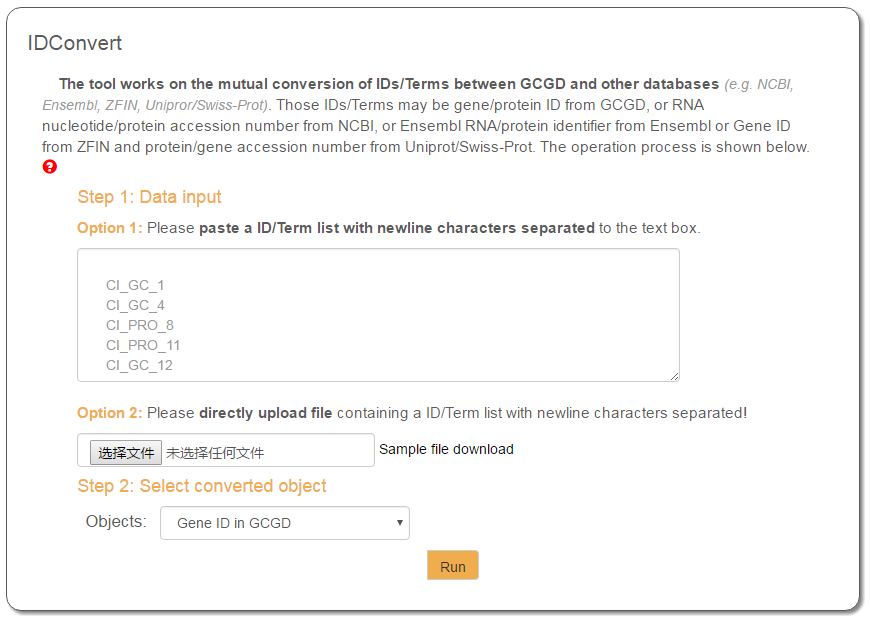
Utilization
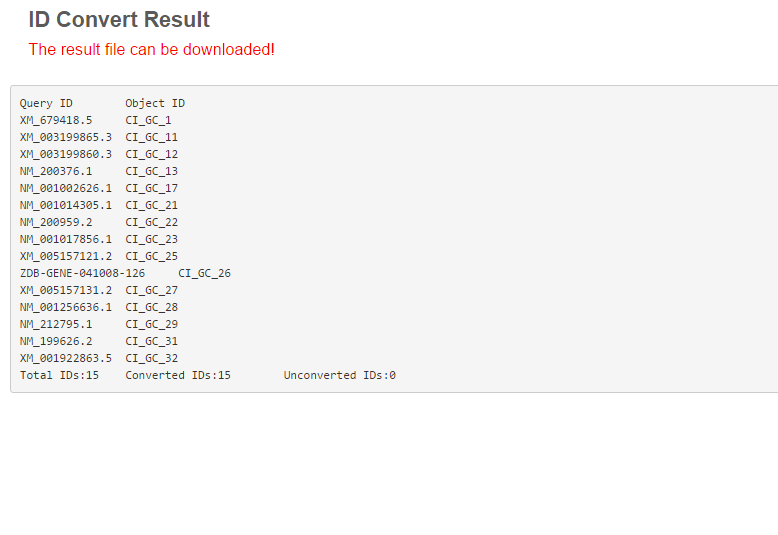
The operation flow of IDConvert contains two steps: the first step is to input data in the text entry, where the user can paste IDs one per row; the second step is to select a database for ID search. All IDs found or not found in the database will be displayed in a new page. Please try the IDConvert page for an experience.
Flowchart
EC2KEGG
EC2KEGG -- a tool for comparison of metabolic pathways between different organisms
General information
EC2KEGG, developed by Aleksey et al. (2014), is an open source Perl package for comparison of metabolic pathways between two organisms. In GCGD, EC2KEGG is adopted to compare the difference of enzymes between the grass carp and some other fishes (e.g., zebrafish, medaka, and southern platyfish, etc.) or human.
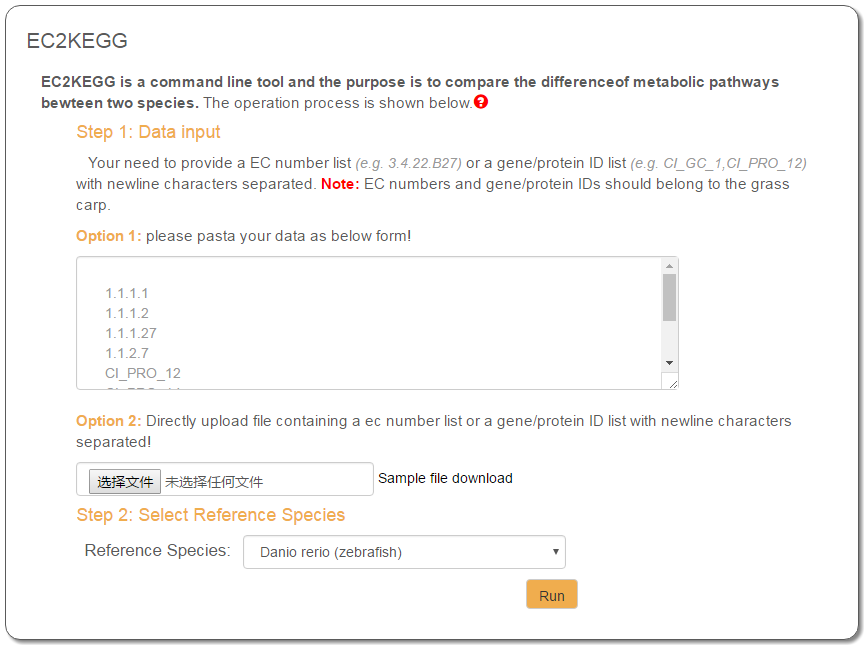
Utilization
The operation processes are presented in the section "EC2KEGG" of the page “Small Tools”. At first, a list of EC numbers (e.g., 3.4.22.B27, 1.1.1.1) or gene/protein IDs (e.g., CI_GC_1, CI_PRO_12), should be input by either pasting in a text entry or uploading as a local file. The second step is to enter a reference organism in a text entry. Currently, GCGD provides the following annotated organisms:
- Zebrafish (Danio rerio)
- Human (Homo sapiens)
- Torafugu (Takifugu rubripes)
- Oryzias latipes (Japanese medaka)
- Southern platyfish (Xiphophorus maculatus)
- Coelacanth (Latimeria chalumnae)
- Zebra mbuna (Maylandia zebra)
- Elephant shark (Callorhinchus milii)
As for other annotated organisms of interest, please see the document for more details.
Result summary
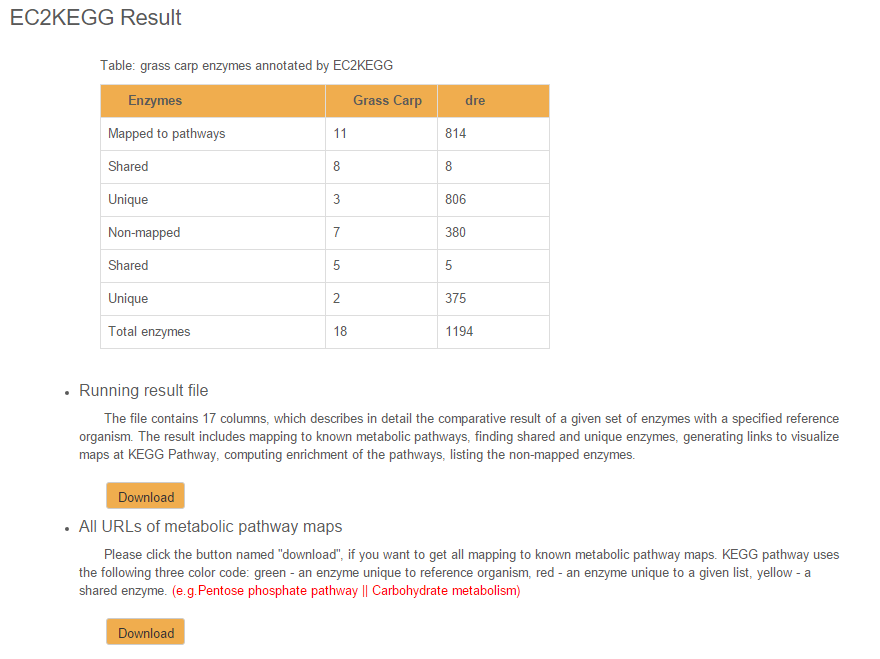
When the "Run" button is clicked, the result of EC2KEGG is displayed in a new page (Fig. 1), which contains two parts: (1) a summary of the searched enzymes in the two species, and (2) result files for download. The main result file reports one KEGG pathway per row, and comprises of 17 columns described as following:
- PathwayID: the ID of the KEGG Pathway
- PathwayName||Category: the name and category of the KEGG pathway
- Total (EC_All): the numbers of all genes known in the pathway
- Total (EC_Ref(xxx)): the number of enzymes found in the reference genome
- Total (EC_Given): the number of enzymes found in the grass carp genome
- Total (EC_Shared): the number of enzymes found in both genomes
- Total (EC_Unique_Ref): the number of enzymes found only in the reference genome
- Total (EC_Unique_Given): the number of enzymes found only in the grass carp genome
- EC_All: all enzymes in the KEGG pathway
- EC_Ref(xxx): enzymes found in the reference genome
- EC_Given: enzymens found in the grass carp genome
- EC_shared: enzymes found in both genomes
- EC_Unique_Ref: enzymes found only in the reference genome
- EC_Unique_Given: enzymes found only in the grass carp genome
- P-value: p-values adjusted to the multiple hypotheses testing using Benjamini and Hochberg correction
- FDR: false discovery rate
- URL: the URL for the KEGG pathway map
Additionally, a file containing all URLs can be downloaded alone.
Note
It may be take a long time to run EC2KEGG, because all information about organism-specific genes, enzymes, and pathways, has to be retrieved from the KEGG database using its new representational state transfer application programming interface (REST API).
Flowchart
Fig. 1 Result of EC2KEGG

Search
General information
Currently, GCGD mainly contains the resources of genome and gene annotations, the genetic linkage maps, microsatellite genetic markers (i.e., Short Sequence Repeats, SSRs), and three transcriptomic datasets under different conditions. To allow users to get access to those data conveniently and quickly, seven search functions are supplied in GCGD, which enable the user to search respectively for
- keyword or a text phrase in the top-right search entry
- SSRs/SNPs used for the construction of the consensus linkage map of grass carp
- SSRs by whole-genome scanning
- Gene annotations
- Gene family
- Similar sequences
- Compound keywords
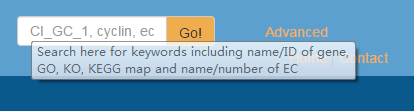
Here, we emphasize how to correctly use the first way, i.e., keyword search in the right-top corner of any page on GCGD. Currently, valid keywords should fall in the types listed below and shown in a floating windows as well (Fig. 1).
- Gene ID/name
- GO ID/name/definitions
- KO ID/name
- EC number/name
- KEGG pathway map number/name
Keyword search returns any gene matches on a result page. The use of other six search functions has been described in detail the corresponding section of the page "Help".
Fig. 1 Keyword search in in the top-right search entry
Data Download
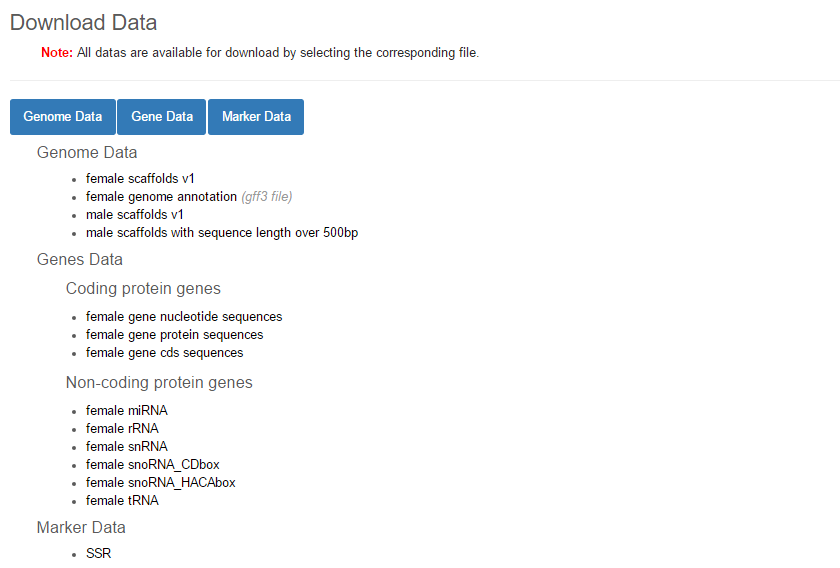
In the Download page, all related genetic and genomic data of grass carp are listed as shown below and can be downloaded by directly clicking on the list.
Others
Please feel free to contact Dr. Xiao-Qin Xia for any questions or comments about the database.