Species-corresponding genomes and annotations
| Species Name | Genome Version | download url |
|---|---|---|
| Danio rerio | GRCz11 | Download |
| Protopterus annectens | GCF_019279795.1 | Download |
| Monopterus albus | GCF_001952655.1 | Download |
| Gadus morhua | GCF_902167405.1 | Download |
| Clupea harengus | GCF_900700415.2 | Download |
| Salmo salar | GCF_000233375.1_ICSASG_V2 | Download |
| Lates calcarifer | ASM164080v1 | Download |
| Collichthys lucidus | GCA_004119915.2 | Download |
| Amia calva | GCA_016984155.1 | Download |
| Ictalurus punctatus | GCF_001660625.1_IpCoco_1.2 | Download |
| Cyprinus carpio | GCF_018340385.1_ASM1834038v1 | Download |
| Perca fluviatilis | GCF_010015445.1 | Download |
| Pimephales promelas | GCF_016745375.1 | Download |
| Ctenopharyngodon idella | GC_v1 | Download |
| Xiphophorus hellerii | GCF_003331165.1 | Download |
| Coregonus clupeaformis | GCF_020615455.1 | Download |
| Larimichthys crocea | GGCF_000972845.2 | Download |
| Siniperca chuatsi | GCF_020085105.1 | Download |
| Oryzias latipes | GCF_002234675.1 | Download |
| Astyanax mexicanus | GCF_023375975.1 | Download |
| Triplophysa tibetana | GCA_008369825.1 | Download |
| Oreochromis niloticus | GCA_001858045.2 | Download |
| Oncorhynchus mykiss | GCF_013265735.2_USDA_OmykA_1.1 | Download |
| Labeo rohita | GCA_004120215.1_ASM412021v1 | Download |
| Hypophthalmichthys molitrix | CNP0000974 | Download |
| Channa argus | GCA_004786185.1 | Download |
| Lepisosteus oculatus | GCF_000242695.1 | Download |
| Tetraodon nigroviridis | GCA_000180735.1 | Download |
| Pangasianodon hypophthalmus | GCF_009078355.1_GENO_Phyp_1.0 | Download |
| Puntigrus tetrazona | GCF_018831695.1 | Download |
| Takifugu rubripes | GCF_901000725.2 | Download |
| Gasterosteus aculeatus | GCF_016920845.1 | Download |
| Nothobranchius fuzeri | GCF_027789165.1 | Download |
| Megalobrama amblycephala | GCF_901000725.2 | Download |
| Tachysurus fulvidraco | GCF_003724035.1_ASM372403v1 | Download |
Gene basic information

Gene basic information: a: Gene basic information, species, chromosome, chromosome position, original database id and description b: Sequence quick pull, select nucleic acid or protein will be presented in fasta format c: Gene compared with Swiss-prot vertebrate data id corresponding information d-e: GO/KEGG annotation information.
RNA information

RNA information: a: Visualization of the gene transcript node, click on the transcript to browse the substructure and download the corresponding sequence b: Visualization of the transcriptome analysis on the gene information page (the process of selecting the data set is similar to the "data selection" in the previous "Omics Online Analysis" " part) c: Statistical test analysis parameter setting d: Analysis result picture display, click "picture ggplot file" below to download the corresponding ggplot2 file, upload it to the Retuning Plots part of Visual Omics, and you can perform online high-freedom degree of picture editing.
Protein information

protein information: a: Prediction and visualization of protein domains corresponding to genes, below are predicted result files, visualization pictures and corresponding ggplot2 files. b: Protein subcellular localization c: Protein interaction information
SNP information
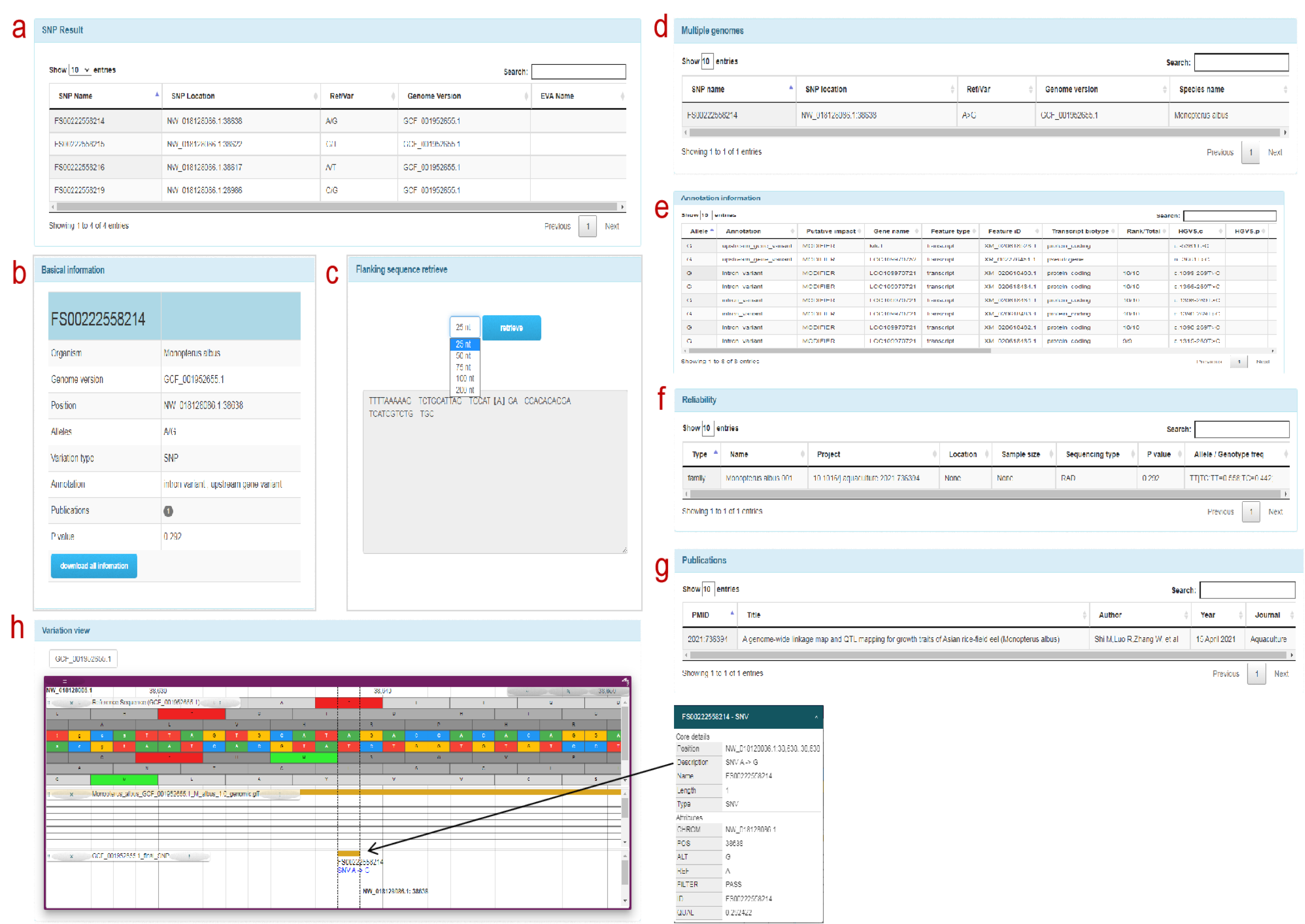
SNP information: a: SNP information associated with the FishCODE gene page, users can click on the SNP form row to jump to the detailed information page of the SNP, b: basic information of the SNP site c: SNP flanking sequence (optional distance) d: multiple genome positions Point comparison information e: SNP genome annotation information. f: Population type, size, sampling location and other information. g: Article information h: SNP visualization information display. Click the target variation to display the position information of the SNP and its adjacent sites on the genome, as well as the detailed information of the SNP site.
Macro related information
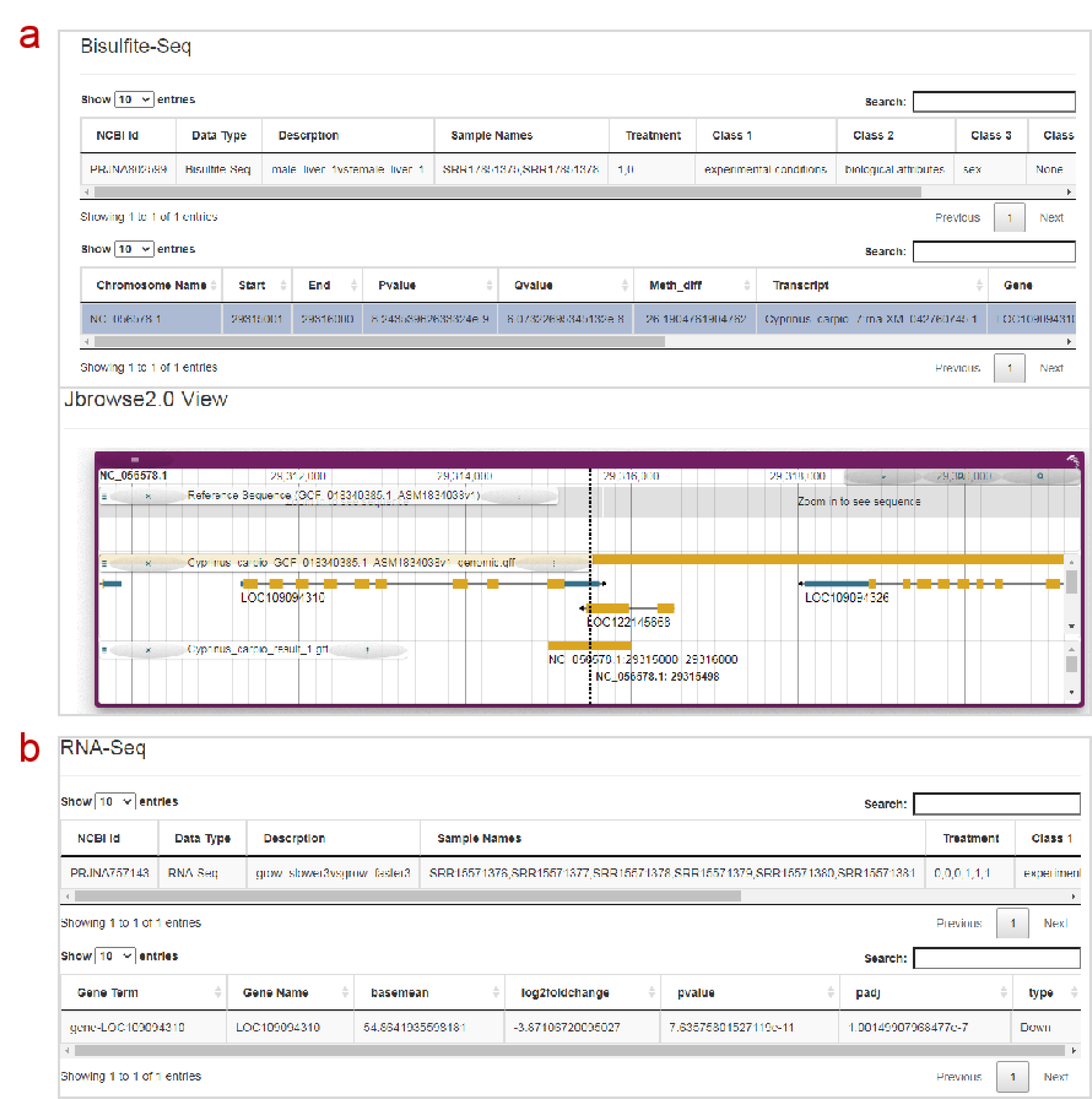
Macro related information: a: Bisulfite-Seq DMRs analyzes the associated gene information, and associates the gene with the experimental conditions of the data set; b: the gene obtained by RNA-Seq differential analysis, associates the gene with the experimental conditions of the data set.
Transcriptome upstream script introduction
The purpose of this part of the code is to help users pre-process their own transcriptome raw data on their own machine. The output of this script is the gene count and TPM file. These output results can be input by the user as an input file to the "upload own data" section of FishCODE's online transcriptome analysis.
Considering the large-scale computing power required for the pre-processing of omics raw data, this series of processing scripts currently only supports Linux or Linux-like systems. More local software supported by the system are in our development list, so please pay attention to our webpage. It is worth noting that although FishCODE focuses on the analysis of fish omics data, this series of scripts is a general omics data processing flow, and users can use this processing flow to process any omics annotations (at least genome and gff files) Species.
Code download
| Version Name | Modify Time | Download | Breif Introdunction |
|---|---|---|---|
| Transcriptome_linux_1.1 | 25 June 2023 | download | The original version, users can use this series of scripts to process omics raw data on the linux system |
Getting started
Before you start
The pp.conf file records the absolute paths of the dependent software mentioned below. Before you start you must configure this file and replace the execution path of the software with the absolute path to the software on your server!
First: create database index for analysis
Dependent software: gffread, salmon 1.4.0, bedtools v2.26.0, mashmap
$ bash First_bulid_index.sh ./build_faidx_example/GCF_901000725.2_fTakRub1.2_genomic.fna ./build_faidx_example/GCF_901000725.2_fTakRub1.2_genomic.gff 30
Note: 1. Your genome and corresponding annotation files must be in the same folder. 2. All the following working directories default to the decompressed directory.
3. The first parameter is the genome location, the second parameter is the gff file location, and the third parameter is the number of threads
Second: quality control
Dependent software: NGSQCToolkit_v2.3.3 (IlluQC.pl)
$ bash filter.sh 792045 SRR17641338
Note: The first parameter is the name of the upper folder where fastq.gz is located, or the project name, and the second parameter is the prefix of fastq.gz. For the specific file structure, please refer to the compressed package. The user must create a project under the parent directory 00_data, and store the sequencing files to be processed under the project name. "./00_data/projectname/*fastq.gz"
Third: count
Dependent software: salmon 1.4.0
$ bash salmon.sh 792045 SRR17641338 ./build_faidx_example/GCF_901000725.2_fTakRub1.2_genomic.fna ./build_faidx_example/GCF_901000725.2_fTakRub1.2_genomic.gff
Note: The first parameter is the project name of the second step, the second parameter is the prefix of fastq.gz, the third parameter is the genome location, and the fourth parameter is the location of the gff file (note that the location of this genome and annotation file must Strictly the input genome and gff file path for the first step)
Others
1. The output result of the third step will be in 01_result/792045/SRR17641338_quant/ under the same directory
2. All directory structures and demonstration data examples are included in the compressed package, allowing users to try it easily.
3. In order to facilitate users to generate transcriptome data corresponding to the species included in FishCODE, we provide a download package of the genome and annotation files of FishCODE's 35 fish species in the "Help->Usage->gene information" section.
Epigenomics upstream script introduction
The purpose of this part of the code is to help users preprocess their own methylome (only referring to the "Bisulfite-Seq" library construction strategy) raw data on their own machines. The output of this script is a file of methylation ratios for the sites. Users can directly import these output results into the methlKit package as input files.
Considering the large-scale computing power required for the pre-processing of omics raw data, this series of processing scripts currently only supports Linux or Linux-like systems. More local software supported by the system are in our development list, so please pay attention to our webpage. It is worth noting that although FishCODE focuses on the analysis of fish omics data, this series of scripts is a general omics data processing flow, and users can use this processing flow to process any omics annotations (at least genome and gff files) Species.
Code download
| Version Name | Modify Time | Download | Breif Introdunction |
|---|---|---|---|
| Methylome_linux_1.1 | 25 June 2023 | download | The original version, users can use this series of scripts to process omics raw data on the linux system |
First: quality control
Dependent software: FASTX Toolkit 0.0.13
$ bash filter_fastx.sh 555065 SRR9697472
Note: The first parameter of the quality control script is the project name, and the second parameter is the prefix name of fastq.gz. But before you run the script, you need to replace "@SRR" on line 20 of "filter_fastx.sh" with the special flag string of your sequencing data. What are special identifiers? Take NCBI's original sequencing file as an example. The special string of SRRXXXX.fastq.gz is "@SRR". You can zless open the fastq.gz file to view the first line and replace the "NCBI's" in line 20 of filter_fastx.sh. @SRR".
Second: count
Dependent software: BSMAP 2.9, samtools 1.3.1, Python 2.7.15 [sys, time, os, array, optparse]
$ bash methy.sh 555065 SRR9697472 ./zebrafish/GCF_000002035.6_GRCz11_genomic.fna
Note: The first parameter of the quality control script is the project name, the second is the prefix name of fastq.gz, and the third parameter is the absolute path of the genome of the corresponding species.
Others
1. The output result of the third step will be in 02methyPos/01methratio/555065 under the same directory
2. All directory structures and demonstration data examples are included in the compressed package, allowing users to try it easily.
3. *methratio.txt is the final output result, where *methratio is the methylation status of all sites, and the final result is the filtered site file with coverage
4. Considering that methylation computing resources consume a lot, we recommend that your running server has at least 100G of RAM, 200G of storage space, and 10 CPU cores. (actually depends on your genome size)
Code download
| Version Name | Modify Time | Download | Breif Introdunction |
|---|---|---|---|
| R_analysis_visualization_linux_1.1 | 25 June 2023 | download | The original version, R code of analysis and visualization |
Dependent R packages
Dependent software:
1.Before the process begins, you need to make sure that the following dependent software in your Linux environment is installed successfully and can be used properly.
please refer FishCODE.yaml.
2.If conda works under your Linux environment, you canimport the corresponding environment.
$ create -f FishCODE.yaml via conda env create -f
3. Enter the corresponding directory and grant execute permission
$ cd R_analysis_visualization
$ chmod 755 ./*R
Transcriptome analysis
$ ./Rversion1.R de_analyse_batch 11 ./data/example_trans/*.data 0 ./data/example_trans/feature > ./data/example_trans/err.txt 2>&1
Time-series transcriptome analysis
$ ./Rversion1.R de_analyse_batch_time 11 ./data/example_time/*.data 0 ./data/example_time/feature > ./data/example_time/err.txt 2>&1
Evolutionary tree construction
$ Rscript ./evolution_tree.R ./data/example_evo/common_aln.phy
SNP annotation
$ ./run_snp2.py -sh ./snpEff_pip1_1.sh -f ./data/example_snp/common.data -i new_grasscarp -c --address xxxxxxxxx@xxx.com -w ./data/example_snp -o ./data/example_snp/out
Note: Considering that SNP annotations need to be built in advance, and that SNP annotations mainly rely on SNPEff software (https://pcingola.github.io/SnpEff), the usage is also relatively simple. This script is mainly used for review and reference.
Methylation analysis
$ Rscript ./methylkit.R SRR17851372,SRR17851373 SRR17851372,SRR17851373 3 1000 1000 0.01 25 all ./data/example_meth/result_1.txt 1,0 10 ./data/example_meth no 0.2 0.01 PRJNA802599,PRJNA802599
Note: Differential methylation analysis.
$ python2 ./find_genes_by_loc.py --gene-file=./Cyprinus_carpio.txt --gene-name=Gene --gene-chr=Chr --gene-start=Start --gene-end=End --loc-chr=chr --loc-start=start --loc-end=stop -i ./data/example_meth/result_1.txt -o ./data/example_meth/SRR17851372,SRR17851373.meth.gene.CG.txt
Annotation of genes directly associated with differentially methylated regions.
Note that you need to use python 2.75 here, not python3.
$ python3 ./find_genes_nearby_loc.py --gene-file=./Cyprinus_carpio.txt --gene-name=Gene --gene-chr=Chr --gene-start=Start --gene-end=End --loc-chr=chr --loc-start=start --loc-end=stop --adjacent=2000 -i ./data/example_meth/SRR17851372,SRR17851373.meth.gene.CG.txt -o ./data/example_meth/SRR17851372,SRR17851373.meth.2000.CG.txt
Note:Annotation of (~2000bp) genes indirectly associated with differentially methylated regions.
Note that you need to use python 2.75 here, not python3.